
Developing new sustainable, selective, and versatile catalysts is one of the big challenges in chemistry. Simulations help to really understand chemical reaction mechanisms by splitting them up into elementary steps. For each step, we calculated the transition state and the barrier. Using that, or the respective rate constants, we combine the elementary steps to an overall turnover frequency, which we can compare to experimental measurements.
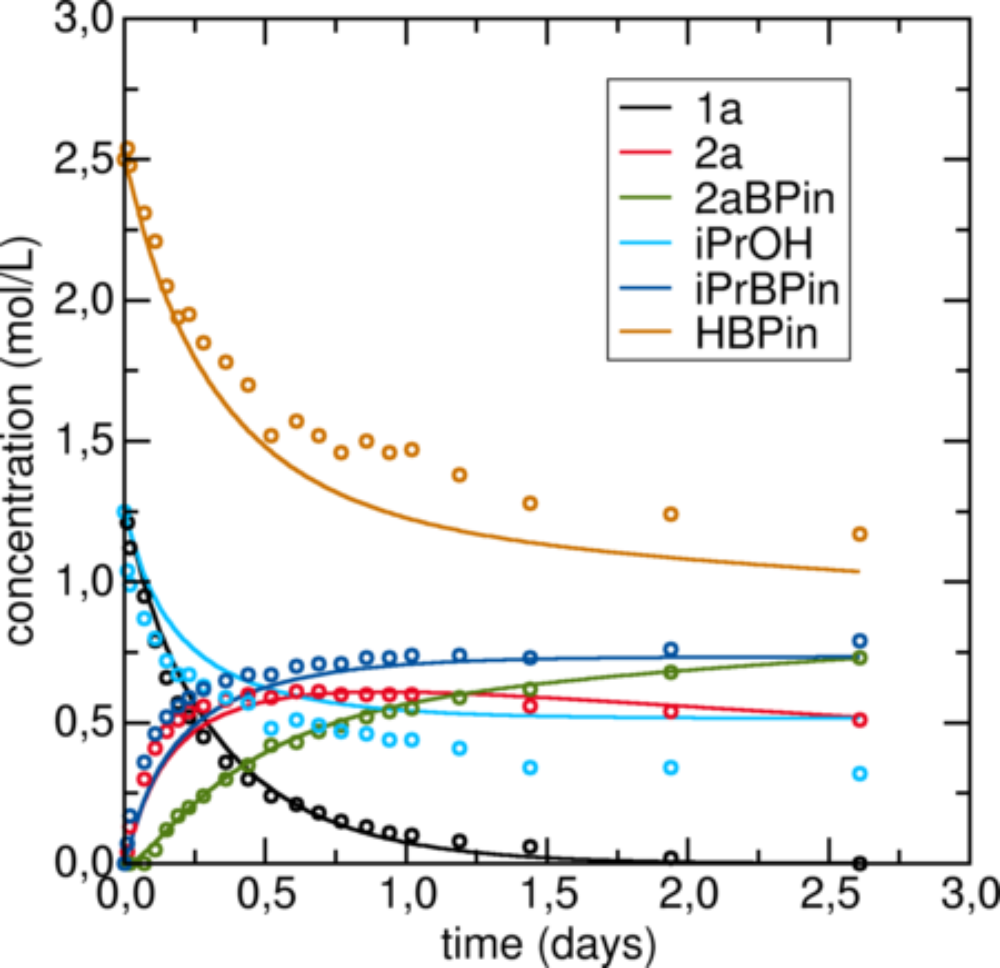
One particularly nice example we looked at was the hydroboration of ketones. Standard DFT with transition state theory was sufficient. However, the product alcohol can also act as one of the reactants, as a proton source. That makes the kinetic model, which describes the overall reaction, somewhat intricate. We had very detailed concentration profiles from our experimental partners, which we could compare with the time-dependent results of our microkinetic model. See the image on the right, or [1] for further details. After we took relevant side reactions into account and understood that they are facilitated by chloride resulting from the activation of the catalyst, we could achieve excellent agreement with the experiments.
We cooperate closely with experimental partners within the collaborative research center CRC 1333 “Molecular Heterogeneous Catalysis in Confined Geometries”. Its goal is to understand and exploit catalysis in mesopores. These pores are large enough to allow substrates to diffuse into the pore but small enough to restrict the movement of the catalytic complex. Within the CRC 1333, we looked at molybdenum-based metathesis, rhodium-catalyzed asymmetric reactions, cooperative catalysis, and several other cases.