
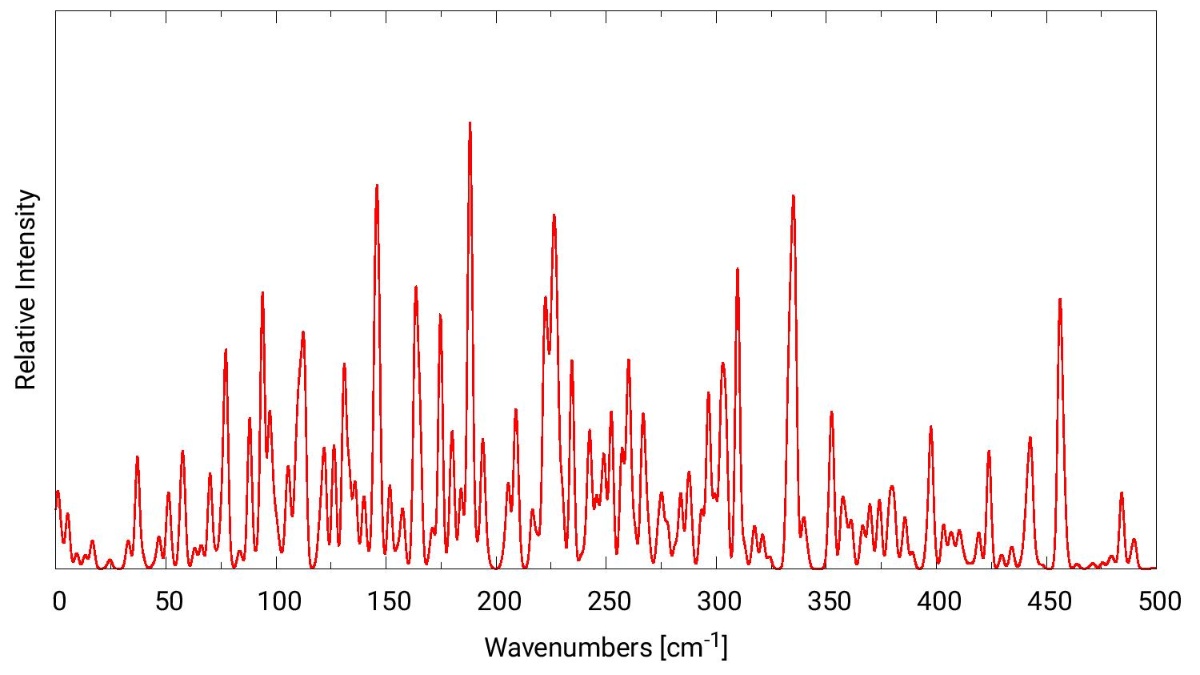
Ab initio calculations of rovibrational spectra of complex organic molecules.
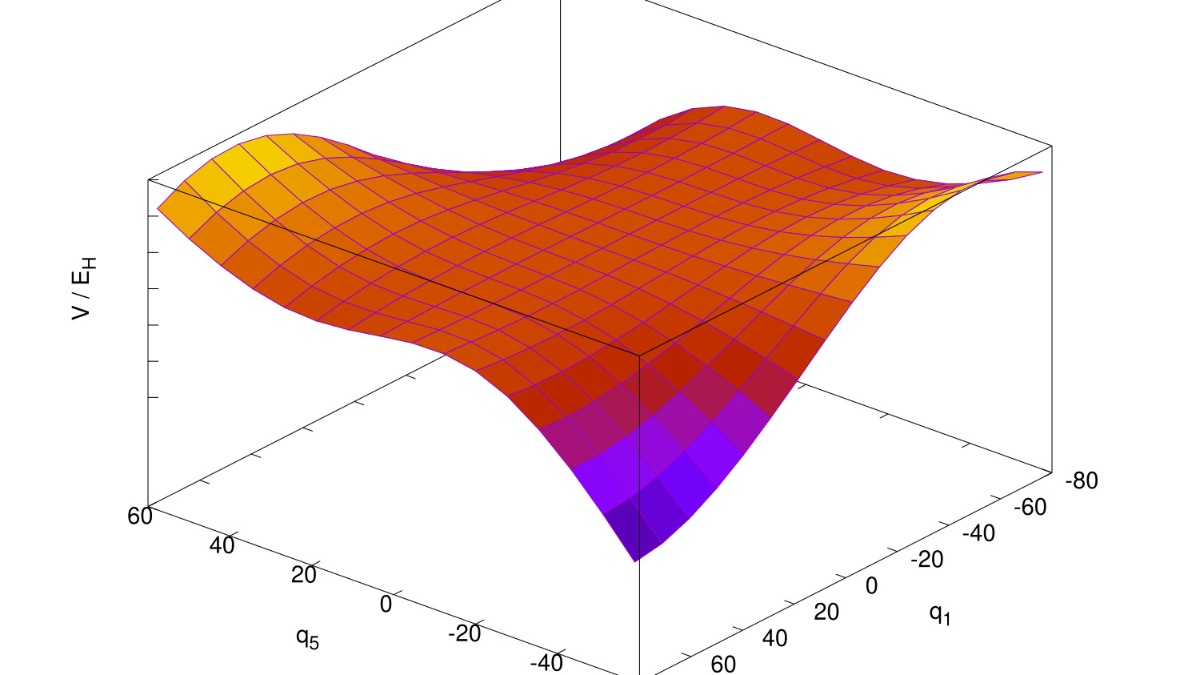
Automated calculation of multidimensional potential energy surfaces.
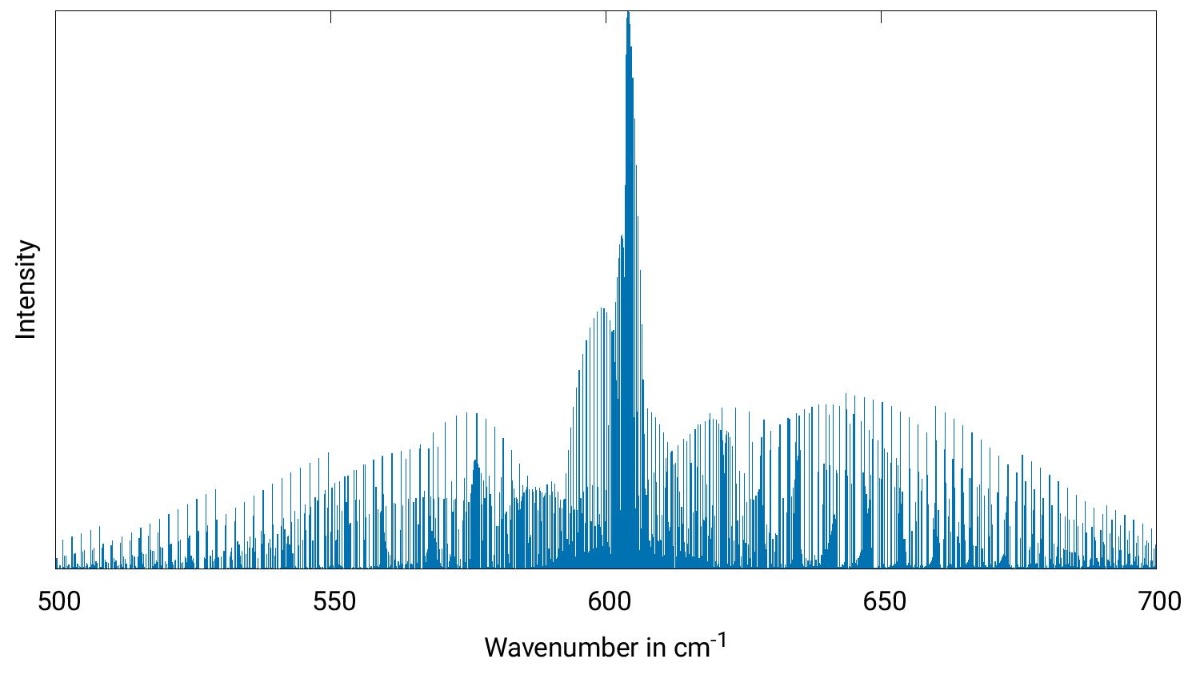
Simulation of Raman microwave spectra.

Spectroscopic investigation of astrochemically relevant molecules.
The theoretical spectroscopy group at the Institute for Theoretical Chemistry focuses on the development and application of highly accurate quantum chemical methods for the calculation of different types of rotational and vibrational spectroscopy. These comprise infrared and Raman spectroscopy, rotational and rovibrational infrared and Raman spectroscopy, but also photoionization and photoelectron spectroscopy. All our program developments enter into the MOLPRO package of ab initio programs, which is used worldwide by many research groups.
Our activities concerning new computational methods aim at automated and user-friendly approaches, which can also be used by non-experts. These developments range from the automated calculation of multidimensional potential energy surfaces, their fitting to analytical representations, the variational calculation of spectra using rovibrational configuration interaction (RVCI) theory, to the implementation of time-independent Raman wavefunction theory for the accurate simulation of photoelectron spectra.
The application of our methods is subject to high-performance computing and we have a number of collaborations with experimentally and theoretically working partners concerning the identification of newly detected molecules and molecular clusters. We are also interested in astrochemistry as our high-level methods can provide reference spectra for molecules being observed in the interstellar medium.