
Our main research focuses on molecules with non-trivial electronic structure, such as radicals, transition metal compounds, and electronically excited molecules.

With ‘non-trivial’ we distinguish these systems from the theoretically more simple case of closed-shell molecules with paired electrons. These are, to zeroth order, well described by a single set of occupied molecular orbitals from which a Slater determinant can be formed (we speak of single-reference systems).
Non-trivial electronic structure means that unpaired electrons are present and that spin-symmetry and static correlation effects have to be considered properly. The latter particularly means that a single set of occupied orbitals is not any more a good zeroth-order description of the system (multireference systems).
We develop new electronic structure methods for describing such multireference systems, with particular focus on coupled-cluster theory. We apply these methods in a number of thrilling research areas, such as photocatalysis, magnetic interactions, electronic energy transfer, and organic semiconductors.
New Developments in Coupled-Cluster Theory
Coupled-cluster theory is the backbone of present-day accurate quantum chemistry. It provides a hierarchy of methods with systematically increasing accuracy, albeit at also increasing computational cost. Coupled-cluster theory allows to make theoretical predictions with definite uncertainty quantification and can be used to provide accurate benchmarks for more approximate methods.
The main challenges in coupled-cluster theory are: (a) The slow converged with basis-set size, due to the interelectronic cusp. (b) The extension to general open-shell and multireference systems. (c) The steep scaling of the computational cost with system size.
Challenge (a) can be addressed by including special basis functions (two-electron functions, also called geminals) that correctly describe the short-range form of the electronic wavefunction. This leads to the field of explicitly correlated approaches, for a review see Chem. Rev. 112, 4 (2012). Here we have contributed to the development of the efficient and accurate approximation CCSD(F12*) [J. Chem. Phys. 123, 231102 (2010)], the question of explicitly correlated triples clusters [J. Chem. Phys. 133, 174118 (2010)] and to explicit correlation of excited states (for a recent work see J. Chem. Phys. 150, 184110 (2019)).
Concerning Challenge (b), it is one of our current main research targets to develop a highly accurate method for complex multireference systems that has the same accuracy as that of CCSD(T) for single-reference molecules. Over the last decade, we made considerable progress in this direction by the development of the internally contracted multireference coupled-cluster method, see J. Chem. Phys. 134, 204111 (2011), J. Chem. Phys. 144, 074103 (2016), Mol. Phys. 118, e1743889.
Challenge (c) has been extensively addressed by the group of Professor Werner and we currently employ these methods in our application projects, see e.g. Eur. J. Org. Chem. 2022, e202101416 (2022). In order to scale up approaches like icMRCC, we have used embedding techniques [J. Chem. Theory Comput. 14, 693 (2018)].
Symbolic Algebra in Quantum Chemistry
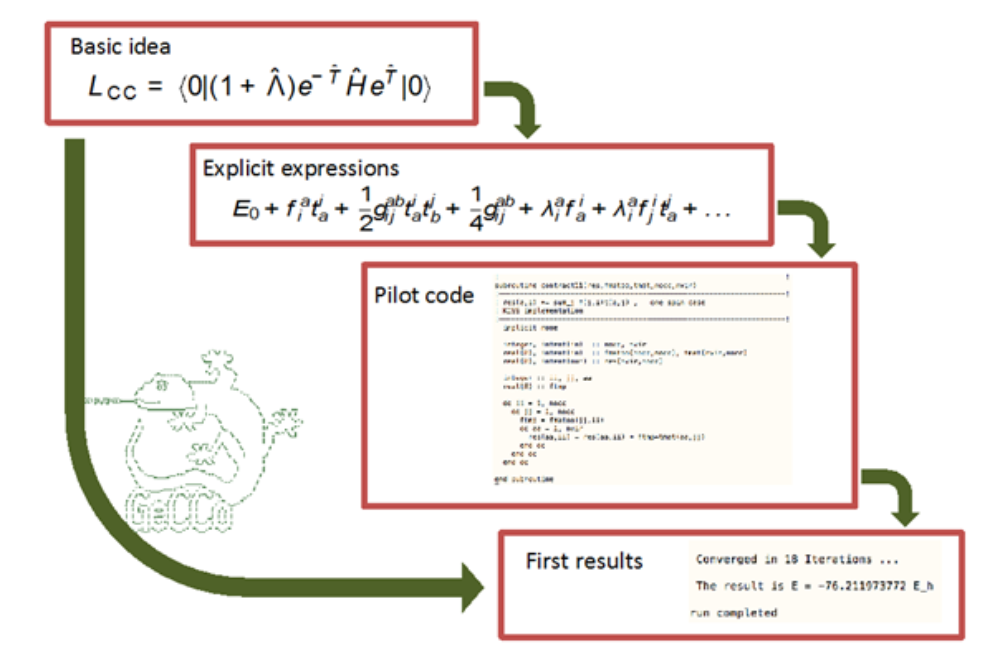
A basic development strategy in our research of new electronic structure methods is automated implemetation. This allows bypassing the otherwise laborious and error-prone “manual” implementation procedure.
The program package GeCCo („General Contraction Code“) encompasses a specialized symbolic algebra part that automates the derivation on the basis of second-quantization, and a numeric part that can evaluate the generated formulae. In particular, this involves the capability of carrying out general tensor contractions.
The program package has in particular been used to implement our multireference coupled-cluster methods. It can be found on GitHub, see github.com/ak-ustutt/GeCCo-public .
Open-shell molecules for (quantum) information processing
Molecules with unpaired electrons offer interesting opportunities for applications. The resulting spin may be used to store information, either classical information (e.g. by encoding bits 0 and 1 as spin-polarization) or even quantum information, by also exploiting quantum superpositions of spin states, thus encoding quantum bits.
From the theory side, the challenge is providing sufficiently accurate ab initio models to parameterize spin Hamiltonian models that are often used to understand the properties of these systems (g tensors, zero-field splitting tensors, hyperfine-coupling tensors). In particular, the processes that drive the decay of the spin-polarized states (spin-lattice relaxation, T1) and the loss of phase information (spin-spin relaxation, T2) are of interest. These are driven by the coupling of the spin to vibrational and (nuclear) spin degrees of freedom in the environment.
Recent contributions comprise a study of spin-orbit operator approximations [J. Chem. Theory Comput. 17, 5530 (2021)], the use of machine learning to approach spin-phonon coupling effects [J. Chem. Theory Comput. 18, 1 (2022)|], the study of bistabilities in non-alternating unsaturated hydrocarbons [Phys. Chem. Chem. Phys. 226, 20462 (2024)], or the study of a biradical bridged dinuclear Ni-complex [J. Inorg. Chem. 63, 6042 (2024)].
Excited Electronic States of Molecules
Even for molecules with a well-behaved closed-shell ground state, the electronic structure in the excited state become more involved as electron pairs are torn apart. Often anti-bonding orbitals become occupied, leading to significant changes of the equilibrium structure in the excited state. Such effects have been investigated by us for a number of cases (e.g. J. Phys. Chem. C 116, 15203 (2012)). We also look into electronic energy transfer (EET, also known as FRET) [J. Chem. Phys. Lett. 5, 262 (2014)], charge transfer [Phys. Chem. Chem. Phys. 16, 20586 (2014)] and couplings of both [J. Chem. Phys. 143, 084106]. We also contribute to the development of methods to treat solvent effects [J. Chem. Theory Comput. 9, 977 (2013)]. Most computations of excited states in our group are based on second-order approximate correlation methods for excited states, like ADC(2) and CC2, which can be viewed as analogues of MP2 for excited states.
More recently we looked at the potential of multireference coupled-cluster methods to provide an accurate treatment and we currently have set out to develop multireference analogues of CC2, which promise more stable results whenever excitations lead to strong weakening of cleavage of bonds, see [J. Chem. Phys. 151, 041106 (2019)] and [J. Chem. Theory Comput. 19, 8671 (2023)].
Visualization
Models that map abstract processes of nature into something that a human brain can understand are of central importance in the natural sciences. In chemistry, three-dimensional models of molecules are central to the understanding of properties and reactivity. Compared to usual screen-based computer graphics, AR/VR setups have a great potential in allowing a much more natural inspection of complex topologies. Our particular vision is immersive parameter space analysis, i.e. the users can modify the structures and inspect molecular properties (like multipole moments, magnetic anisotropies) as a function of molecular structure.
Central to our efforts is the program package chARpack (chemistry in Augmented Reality package). For more information please visit the project home page or the GitHub repository.
The project is a close interaction with the group of Prof. Michael Sedlmair from VISUS within SimTech and the Cluster of Excellence EXC2075.
Recent publications:
chARpack: The Chemistry Augmented Reality Package
Understanding Collaborative Learning of Molecular Structures in AR with Eye Tracking
